Chemical synthesis yields potential antibiotic
A new strategy for producing a natural compound could also be used to generate variants with even stronger antimicrobial activity.
Chemists at MIT have developed a novel way to synthesize himastatin, a natural compound that has shown potential as an antibiotic.
Using their new synthesis, the researchers were able not only to produce himastatin but also to generate variants of the molecule, some of which also showed antimicrobial activity. They also discovered that the compound appears to kill bacteria by disrupting their cell membranes. The researchers now hope to design other molecules that could have even stronger antibiotic activity.
“What we want to do right now is learn the molecular details about how it works, so we can design structural motifs that could better support that mechanism of action. A lot of our effort right now is to learn more about the physicochemical properties of this molecule and how it interacts with the membrane,” says Mohammad Movassaghi, an MIT professor of chemistry and one of the senior authors of the study.
Brad Pentelute, an MIT professor of chemistry, is also a senior author of the study, which appears today in Science. MIT graduate student Kyan D’Angelo is the lead author of the study, and graduate student Carly Schissel is also an author.
Mimicking nature
Himastatin, which is produced by a species of soil bacteria, was first discovered in the 1990s. In animal studies, it was found to have anticancer activity, but the required doses had toxic side effects. The compound also showed potential antimicrobial activity, but that potential hasn’t been explored in detail, Movassaghi says.
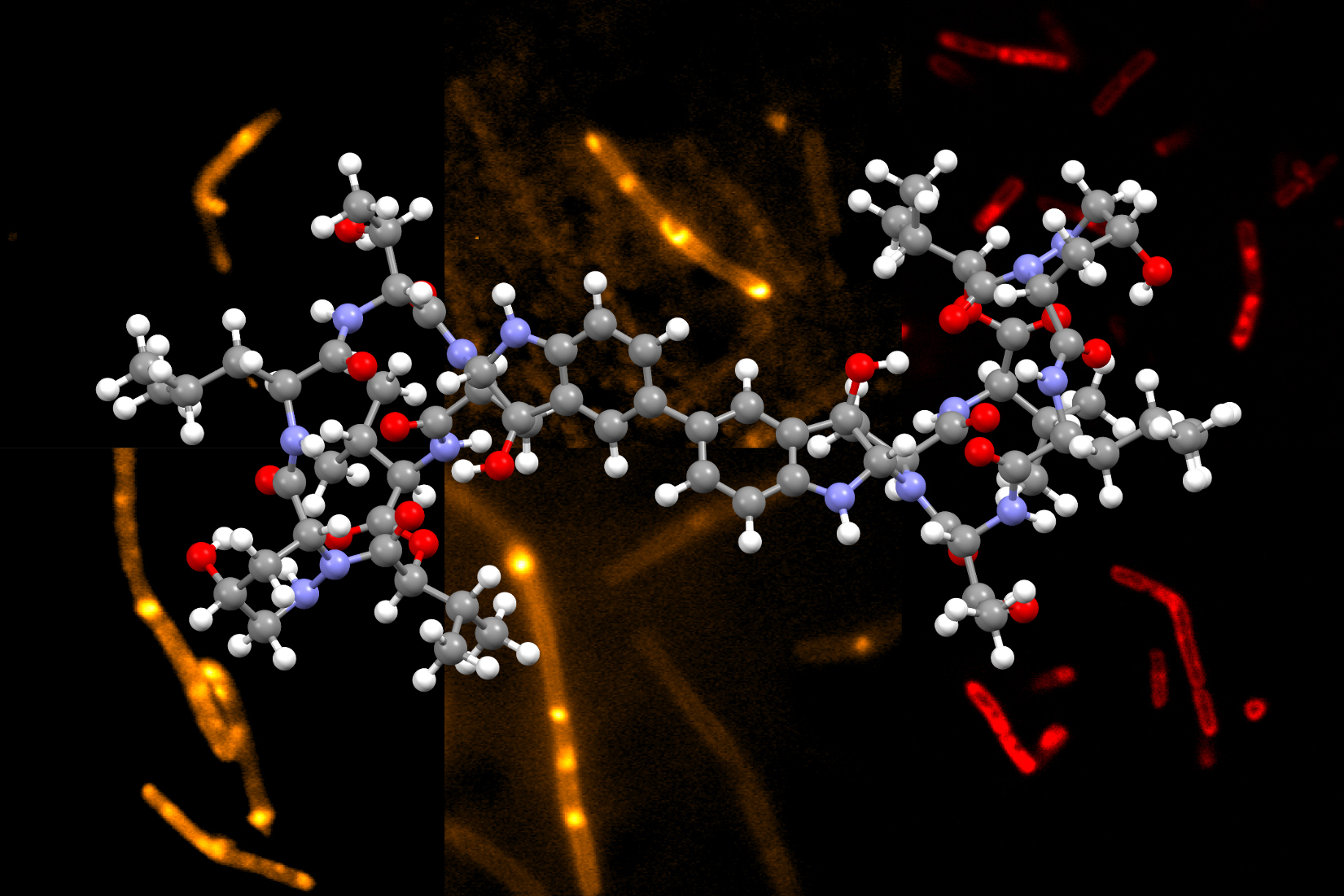
Himastatin is a complex molecule that consists of two identical subunits, known as monomers, that join together to form a dimer. The two subunits are hooked together by a bond that connect a six-carbon ring in one of the monomers to the identical ring in the other monomer.
This carbon-carbon bond is critical for the molecule’s antimicrobial activity. In previous efforts to synthesize himastatin, researchers have tried to make that bond first, using two simple subunits, and then added more complex chemical groups onto the monomers.
The MIT team took a different approach, inspired by the way this reaction is performed in bacteria that produce himastatin. Those bacteria have an enzyme that can join the two monomers as the very last step of the synthesis, by turning each of the carbon atoms that need to be joined together into highly reactive radicals.
To mimic that process, the researchers first built complex monomers from amino acid building blocks, helped by a rapid peptide synthesis technology previously developed by Pentelute’s lab.
“By using solid-phase peptide synthesis, we could fast-forward through many synthetic steps and mix-and-match building blocks easily,” D’Angelo says. “That’s just one of the ways that our collaboration with the Pentelute Lab was very helpful.”
The researchers then used a new dimerization strategy developed in the Movassaghi lab to connect two complex molecules together. This new dimerization is based on the oxidation of aniline to form carbon radicals in each molecule. These radicals can react to form the carbon-carbon bond that hooks the two monomers together. Using this approach, the researchers can create dimers that contain different types of subunits, in addition to naturally occurring himastatin dimers.
“The reason we got excited about this type of dimerization is because it allows you to really diversify the structure and access other potential derivatives very quickly,” Movassaghi says.
Membrane disruption
One of the variants that the researchers created has a fluorescent tag, which they used to visualize how himastatin interacts with bacterial cells. Using these fluorescent probes, the researchers found that the drug accumulates in the bacterial cell membranes. This led them to hypothesize that it works by disrupting the cell membrane, which is also a mechanism used by at least one FDA-approved antibiotic, daptomycin.
The researchers also designed several other himastatin variants by swapping in different atoms in specific parts of the molecule, and tested their antimicrobial activity against six bacterial strains. They found that some of these compounds had strong activity, but only if they included one naturally occurring monomer along with one that was different.
“By bringing two complete halves of the molecule together, we could make a himastatin derivative with only a single fluorescent label. Only with this version could we do microscopy studies that offered evidence of himastatin’s localization within bacterial membranes, because symmetric versions with two labels did not have the right activity,” D’Angelo says.
Andrew Myers, a professor of chemistry at Harvard University, says that the new synthesis features “fascinating new chemical innovations.”
“This approach permits oxidative dimerization of fully synthetic monomer subunits to prepare the antibiotic himastatin, in a manner related to its biosynthesis,” says Myers, who was not involved in the research. “By synthesizing a number of analogs, important structure-activity relationships were revealed, as well as evidence that the natural product functions at the level of the bacterial envelope.”
The researchers now plan to design more variants that they hope might have more potent antibiotic activity.
“We’ve already identified positions that we can derivatize that could potentially either retain or enhance the activity. What’s really exciting to us is that a significant number of the derivatives that we accessed through this design process retain their antimicrobial activity,” Movassaghi says.
The research was funded by the National Institutes of Health, the Natural Sciences and Engineering Research Council of Canada, and a National Science Foundation graduate research fellowship.