Kulik paper published in Nature Computational Science
The paper, A transferable recommender approach for selecting the best density functional approximations in chemical discovery, was published on 12/22/22.
Computational methods are speedy tools needed to discover materials from a large space of all possible materials. When discovering new materials, it’s hard to know which computational method will be accurate. In a recently published paper, members of Professor Heather J. Kulik‘s group describe their development of an artificial intelligence model that can make these decisions for researchers, telling them which method will achieve predictive accuracy needed to guide experiments.
A transferable recommender approach for selecting the best density functional approximations in chemical discovery
Chenru Duan, Aditya Nandy, Ralf Meyer, Naveen Arunachalam, and Heather J. Kulik
Nature Computational Science, 22 December, 2022
DOI: 10.1038/s43588-022-00384-0
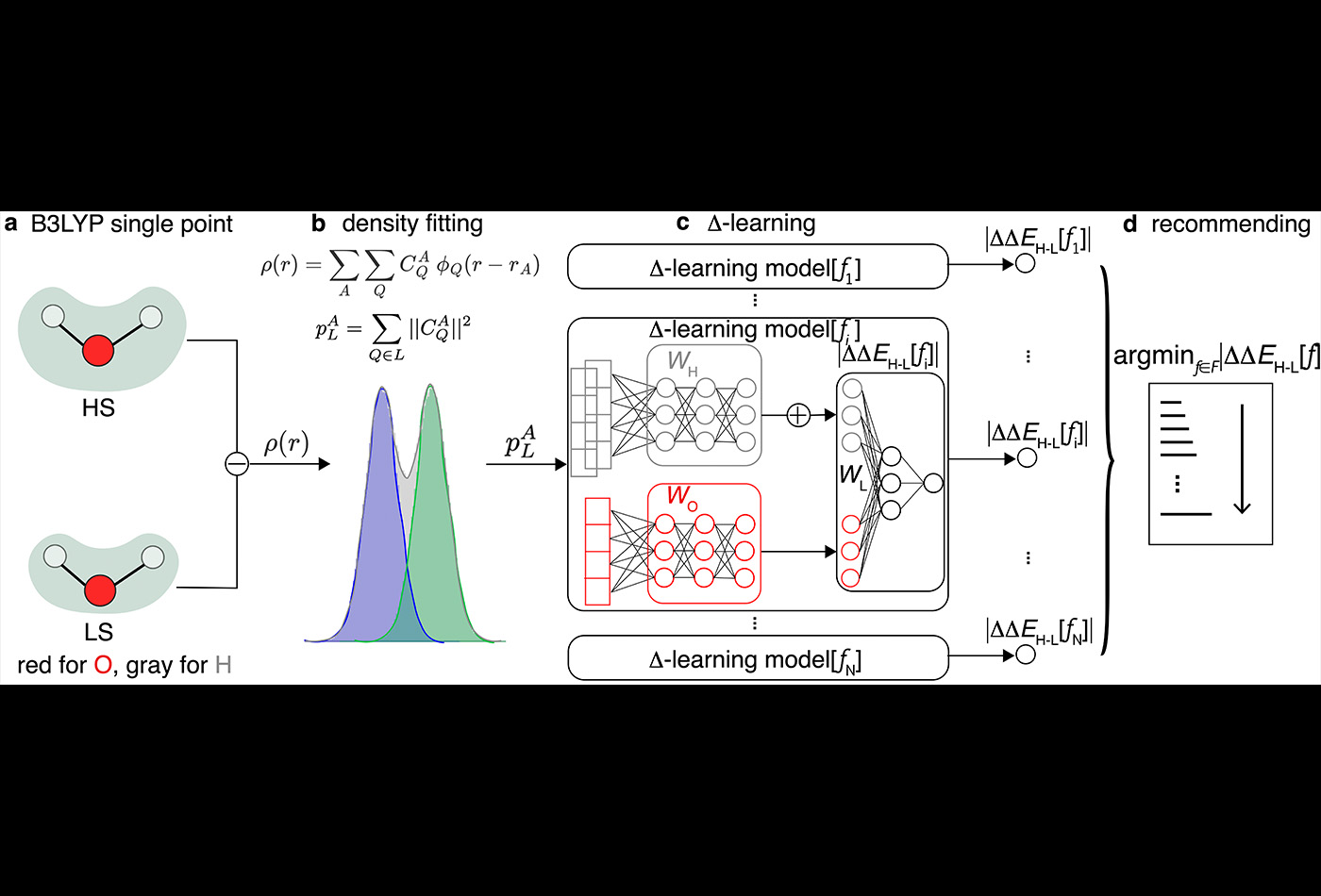
Abstract: Approximate density functional theory has become indispensable owing to its balanced cost–accuracy trade-off, including in large-scale screening. To date, however, no density functional approximation (DFA) with universal accuracy has been identified, leading to uncertainty in the quality of data generated from density functional theory. With electron density fitting and Δ-learning, we build a DFA recommender that selects the DFA with the lowest expected error with respect to the gold standard (but cost-prohibitive) coupled cluster theory in a system-specific manner. We demonstrate this recommender approach on the evaluation of vertical spin splitting energies of transition metal complexes. Our recommender predicts top-performing DFAs and yields excellent accuracy (about 2 kcal mol−1) for chemical discovery, outperforming both individual Δ-learning models and the best conventional single-functional approach from a set of 48 DFAs. By demonstrating transferability to diverse synthesized compounds, our recommender potentially addresses the accuracy versus scope dilemma broadly encountered in computational chemistry.
Read the Full Text at Nature Computational Science.
The Kulik Group leverages multi-scale modeling, electronic structure calculations, and machine learning for the discovery of new molecules and mechanisms in a range of materials from metal-organic frameworks to enzymes and organometallics.