A new computational framework illuminates the hidden ecology of diseased tissues
MESA uses ecological theory to map cellular diversity and spatial patterns in tissues, offering new insights into disease progression.
To understand what drives disease progression in tissues, scientists need more than just a snapshot of cells in isolation—they need to see where the cells are, how they interact, and how that spatial organization shifts across disease states. A new computational method called MESA (Multiomics and Ecological Spatial Analysis) detailed in a study published in Nature Genetics is helping researchers study diseased tissues in more meaningful ways.
The work details the results of a collaboration between researchers from MIT, Stanford University, Weill Cornell Medicine, the Ragon Institute of MGH, MIT, and Harvard, and the Broad Institute, and was led by the Stanford team.
MESA brings an ecology-inspired lens to tissue analysis. It offers a pipeline to interpret spatial omics data—the product of cutting-edge technology that captures molecular information along with the location of cells in tissue samples. These data provide a high-resolution map of tissue “neighborhoods,” and MESA helps make sense of the structure of that map.
“By integrating approaches from traditionally distinct disciplines, MESA enables researchers to better appreciate how tissues are locally organized and how that organization changes in different disease contexts, powering new diagnostics and the identification of new targets for preventions and cures,” says Alex K. Shalek, the Director of the Institute for Medical Engineering & Science (IMES), the J. W. Kieckhefer Professor in IMES & Chemistry, and an extramural member of the Koch Institute at MIT, as well as an Institute Member of the Broad Institute and a Member of the Ragon Institute.
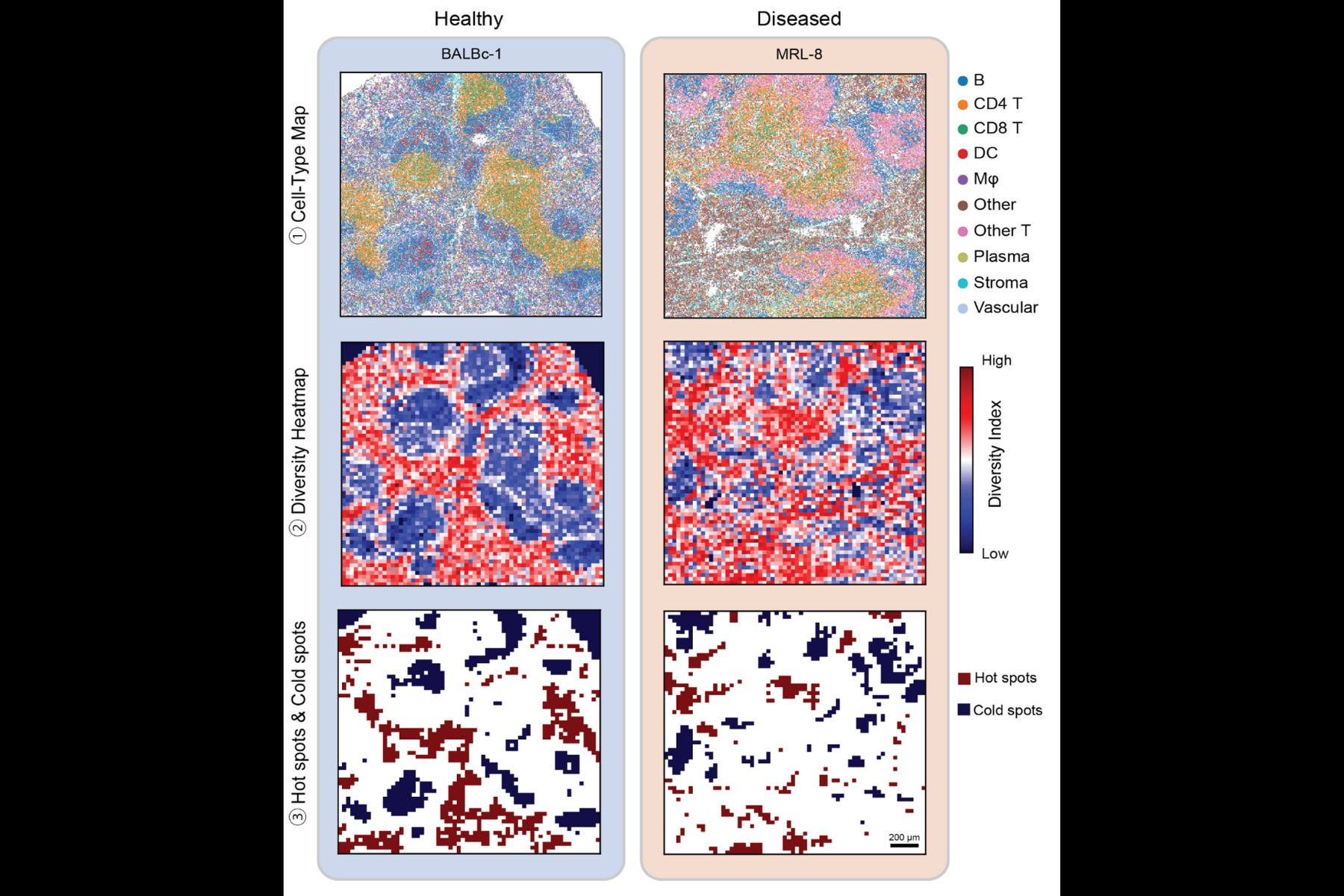
“In ecology, people study biodiversity across regions—how animal species are distributed and interact,” explains Bokai Zhu, MIT postdoc and author on the study. “We realized we could apply those same ideas to cells in tissues. Instead of rabbits and snakes, we analyze T cells and B cells.”
By treating cell types like ecological species, MESA quantifies “biodiversity” within tissues and tracks how that diversity changes in disease. For example, in liver cancer samples, the method revealed zones where tumor cells consistently co-occurred with macrophages, suggesting these regions may drive unique disease outcomes.
“Our method reads tissues like ecosystems, uncovering cellular ‘hotspots’ that mark early signs of disease or treatment response,” Zhu adds. “This opens new possibilities for precision diagnostics and therapy design.”
MESA also offers another major advantage: it can computationally enrich tissue data without the need for more experiments. Using publicly available single-cell datasets, the tool transfers additional information—such as gene expression profiles—onto existing tissue samples. This approach deepens understanding of how spatial domains function, especially when comparing healthy and diseased tissue.
In tests across multiple datasets and tissue types, MESA uncovered spatial structures and key cell populations that were previously overlooked. It integrates different types of omics data, such as transcriptomics and proteomics, and builds a multi-layered view of tissue architecture.
Currently available as a Python package, MESA is designed for academic and translational research. Although spatial omics is still too resource intensive for routine in-hospital clinical use, the technology is gaining traction among pharmaceutical companies, particularly for drug trials where understanding tissue responses is critical.
“This is just the beginning,” says Zhu. “MESA opens the door to using ecological theory to unravel the spatial complexity of disease—and ultimately, to better predict and treat it.”